In this tutorial, we use TopPIC suite to analyze two top-down MS/MS data files on a computer with a Windows 10 operating system. Annotated proteoform spectrum matches (PrSMs) identified by TopPIC from the data files can be browsed here.
1. Overview
2. TopFD
Create the folders below for software packages and data sets used in this tutorial.
- Create a new folder named toppic_tutorial on the C: drive of your system.
- Create a new subfolder named toppic in the folder C:\toppic_tutorial\ for the software TopPIC suite.
- Create a new subfolder named tutorial_1 in the folder C:\toppic_tutorial\.
- Create a new subfolder named tutorial_2 in the folder C:\toppic_tutorial\.
- Create a new subfolder named tutorial_3 in the folder C:\toppic_tutorial\.
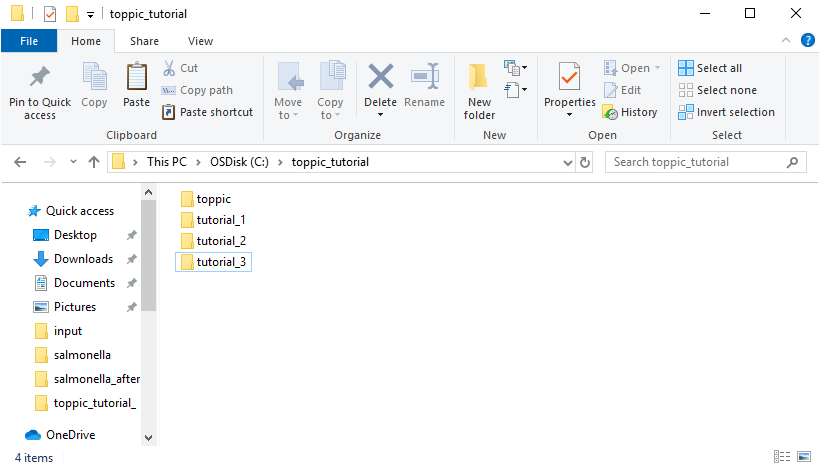
The resulting folder structure is shown in the screenshot below.
3. Software tools
3.1 Msconvert
Msconvert is a software tool in ProteoWizard that converts raw files into various spectrum file formats.
3.1.1 Required software packages to install ProteoWizard
Microsoft .NET Framework 4.0 or a higher version is required. If you are using Windows 10, Microsoft .NET Framework 4.0 has been preinstalled.
3.1.2 Download ProteoWizard
Follow the steps below to download ProteoWizard:
- Go to the link ProteoWizard.
- Choose the type 'Windows 64-bit installer' for end users and download ProteoWizard.
- Double click the downloaded file pwiz-setup-3.0-x86-64.msi to install it.
3.2 TopPIC suite
3.2.1 Download TopPIC suite
- Go to the download webpage of TopPIC suite.
- Choose the download type "Windows 64-bit zip file", fill out the registration form, and click "I accept license agreement and download TopPIC suite" to download it.
- Save it to the folder C:\toppic_tutorial\toppic\.
- Extract all the files of the downloaded zip file to the folder C:\toppic_tutorial\toppic\.
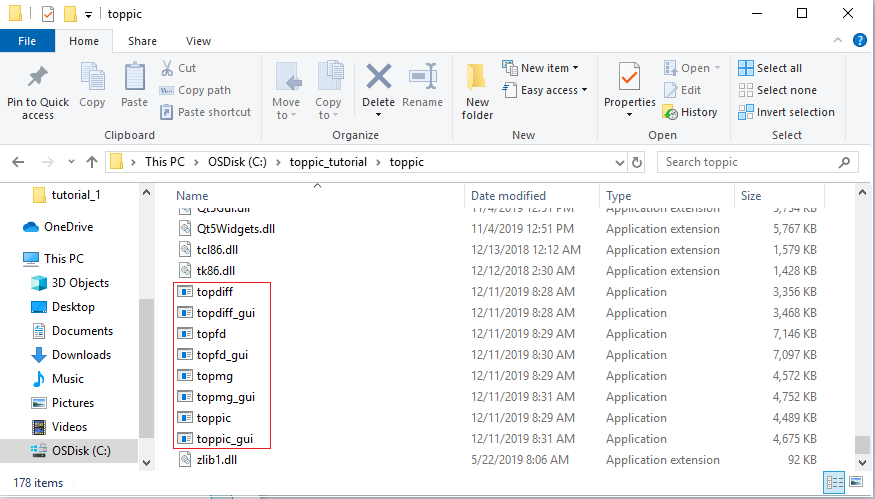
4. Tutorial 1
In this tutorial, we will use TopFD and TopPIC to analyze a top-down MS/MS data set of Salmonella typhimurium for proteoform identification.
4.1 Top-down MS/MS Date set
In the MS experiment, the protein extract of S. typhimurium was reduced with dithiothreitol and alkylated with iodoacetamide. The protein mixture was first separated by gas-phase fractionation, resulting in 7 fractions. Each fraction was separated by an HPLC system coupled to an LTQ-Orbitrap mass spectrometer (Thermo Fisher Scientific). MS and MS/MS spectra were collected at a resolution of 60,000 and 30,000, respectively. In this tutorial, we use only the data files of two fractions (st_1.raw and st_2.raw).
Click here to download the data set, save it in the folder C:\toppic_tutorial\tutorial_1\, and unzip it in the same folder.
4.2 Protein sequence database
A S. typhimurium proteome database of 1,799 proteins was downloaded from the UniProt database.
Click here to download the protein database and save it in the folder C:\toppic_tutorial\tutorial_1\.
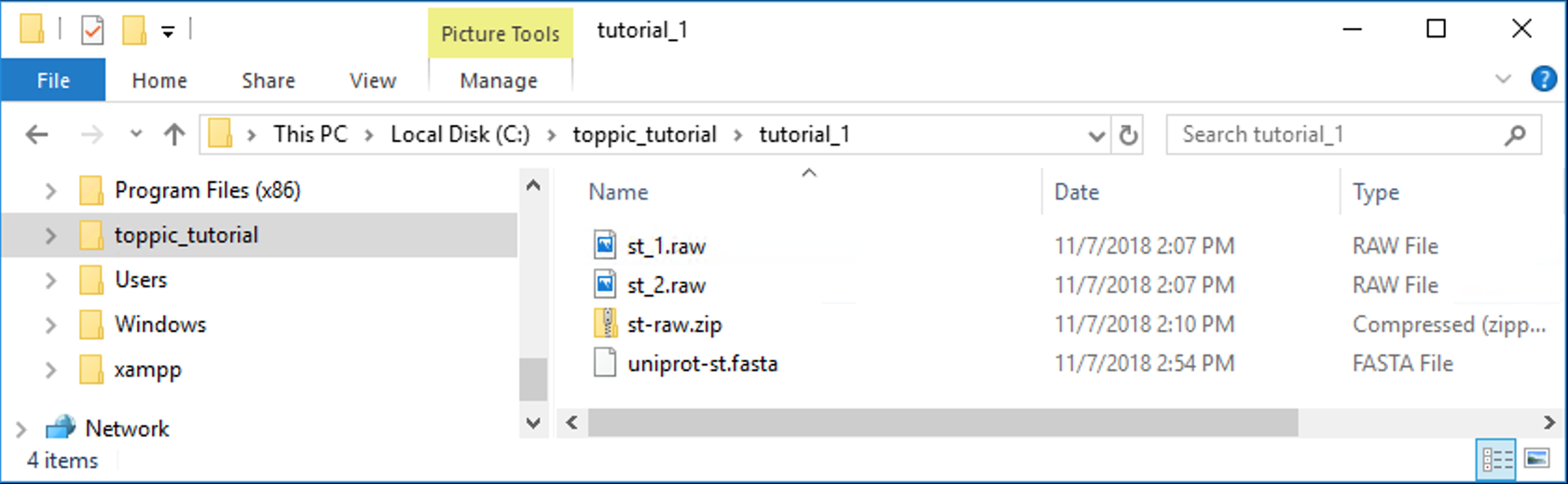
The folder C:\toppic_tutorial\tutorial_1\ is shown in the screenshot below.
4.3 File format conversion
We use MSConvertGUI to convert the raw files st_1.raw and st_2.raw to mzML files.
- Search "msconvert" in the search box on the task bar of Windows 10, and run the desktop app "MSConvert."
- Add the files c:\toppic_tutorial\tutorial_1\st_1.raw and c:\toppic_tutorial\tutorial_1\st_2.raw as input files.
- Add the filter "peakPeaking vendor msLevel=1-" (important).
- Click to the button "Start" to perform file format conversion.
The screenshot of MSConvertGUI is shown below.

In the above file format conversion, the peak picking filter (step 3) is used to generate centroid, not profile, mzML data files, which are required by the spectral deconvolution tool TopFD.
The resulting mzML files are
C:\toppic_tutorial\tutorial_1\st_1.mzML
and
C:\toppic_tutorial\tutorial_1\st_2.mzML
The sizes of the two files are about 41 MB and 47 MB, respectively. They can be downloaded here. The running time for the file format conversion is less than one minute.
4.4 Mass spectral deconvolution
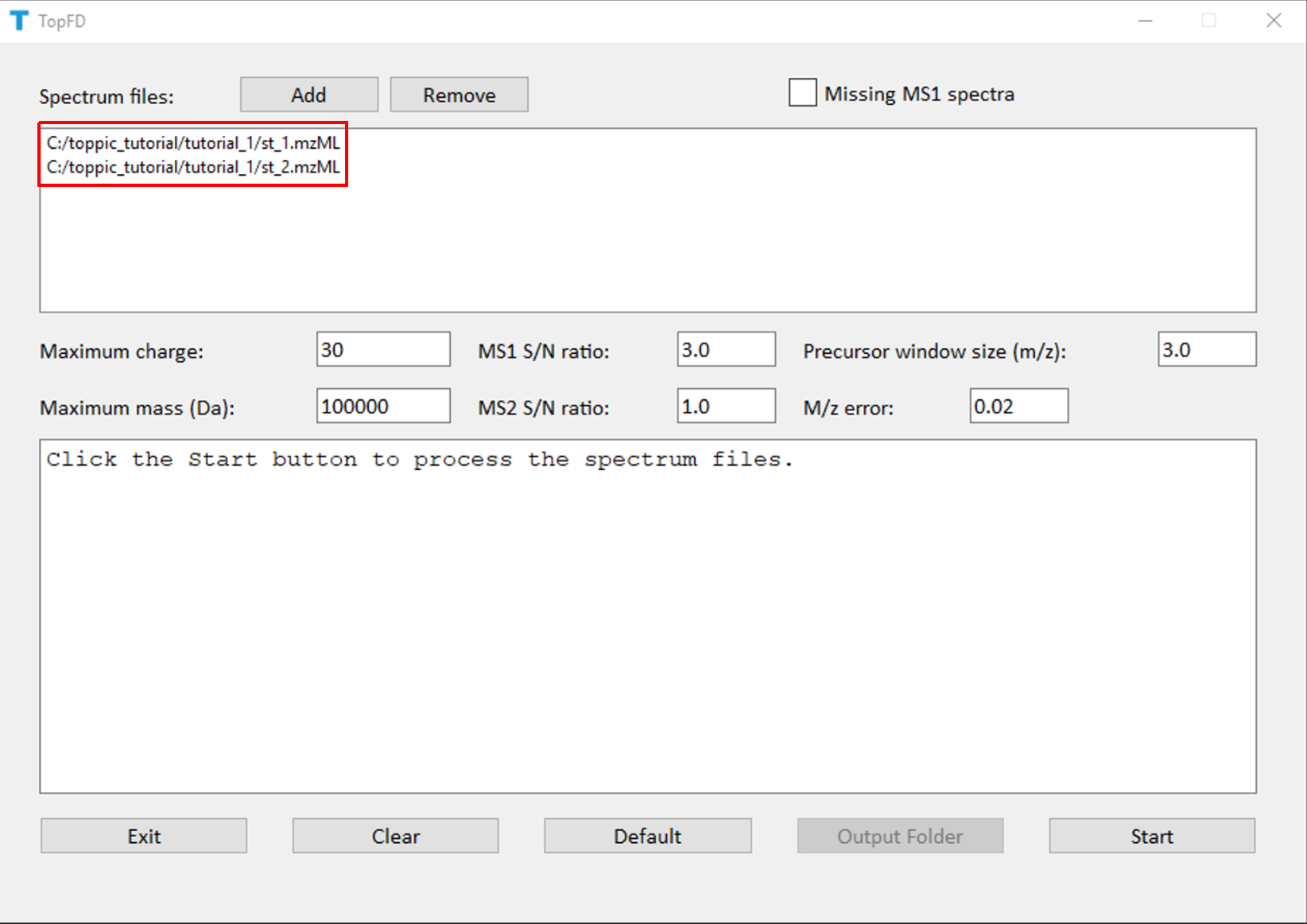
We use topfd_gui for top-down mass spectral deconvolution.
- Double click the executable file topfd_gui.exe in the folder C:\toppic_tutorial\toppic.
- Add the file c:\toppic_tutorial\tutorial_1\st_1.mzML and c:\toppic_tutorial\tutorial_1\st_2.mzML as input files.
- Click to the button "Start" to deconvolute the file.
The screenshot of topfd_gui is shown below.
TopFD reports eight text files and four folders.
- Two msalign files containing deconvoluted MS/MS spectra: C:\toppic_tutorial\tutorial_1\st_1_ms2.msalign
C:\toppic_tutorial\tutorial_1\st_2_ms2.msalign - Four text files containing LC/MS features: C:\toppic_tutorial\tutorial_1\st_1_ms1.feature
C:\toppic_tutorial\tutorial_1\st_1_ms2.feature
C:\toppic_tutorial\tutorial_1\st_2_ms1.feature
C:\toppic_tutorial\tutorial_1\st_2_ms2.feature - Two xml files containing LC/MS features: C:\toppic_tutorial\tutorial_1\st_1_feature.xml
C:\toppic_tutorial\tutorial_1\st_2_feature.xml - Two folders containing deconvoluted MS/MS spectra in the java script format C:\toppic_tutorial\tutorial_1\st_1_html
C:\toppic_tutorial\tutorial_1\st_2_html - Two folders containing a msalign file for MS1 spectra and a csv file for its mzRT features C:\toppic_tutorial\tutorial_1\st_1_file
C:\toppic_tutorial\tutorial_1\st_2_file
The output files and folders can be downloaded here.
4.5 Mass spectral identification using TopPIC
We use toppic_gui to search the MS/MS spectra in st_1_ms2.msalign and st_2_ms2.msalign against the protein database uniprot-st.fasta to identify PrSMs.
- Double click the executable file toppic_gui.exe in the folder C:\toppic_tutorial\toppic.
- Select C:\toppic_tutorial\tutorial_1\uniprot-st.fasta as the protein database file.
- Add C:\toppic_tutorial\tutorial_1\st_1_ms2.msalign and C:\toppic_tutorial\tutorial_1\st_2_ms2.msalign as mass spectrum data files.
- Input "combined" as the file name for combined identifications.
- Select C57 as the fixed modification.
- Check the checkbox Decoy database.
- Select FDR as the spectrum level cutoff type.
- Select FDR as the proteoform level cutoff type.
- Click to the button "Start"
The screenshots of toppic_gui are shown below.
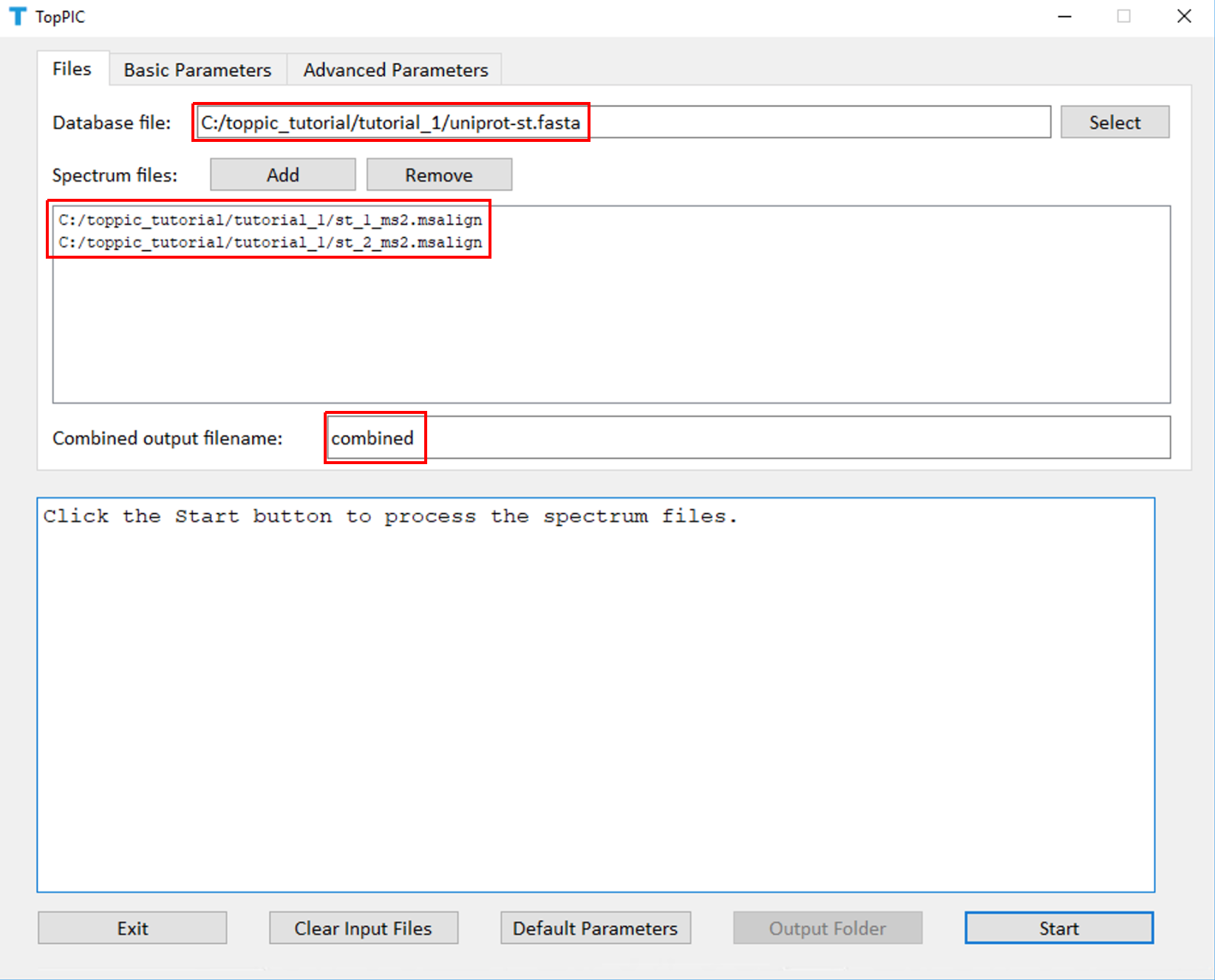
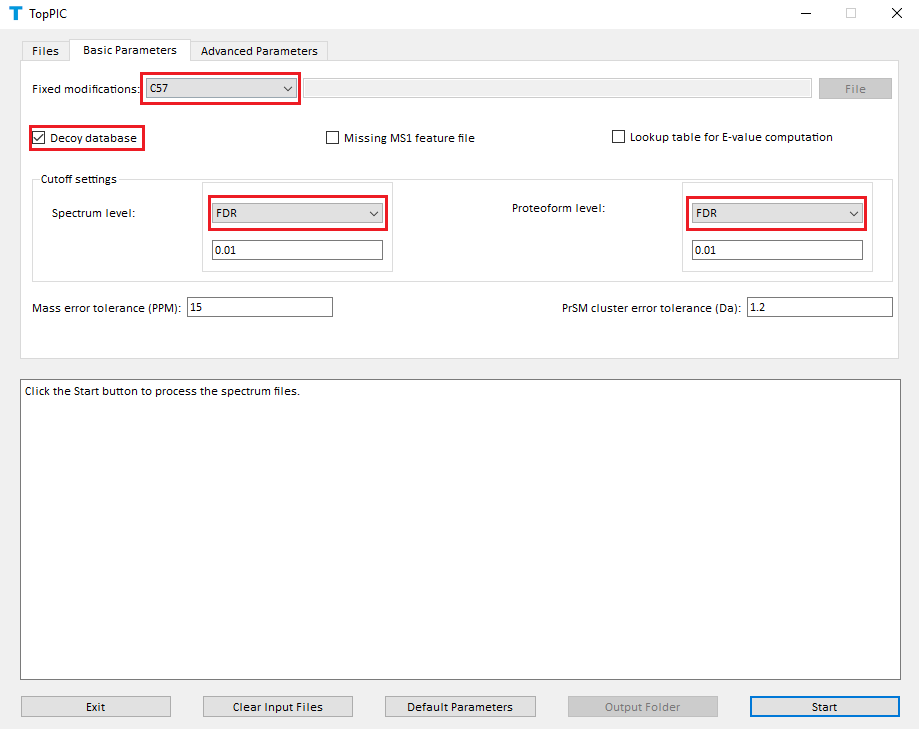
For each input msalign file, TopPIC reports two csv files, an xml file, and collections of html files for identified proteoforms. For example, the output files for st_1_ms2.msalign are
- A csv file containing identified PrSMs with a 1% spectrum level FDR: C:\toppic_tutorial\tutorial_1\st_1_ms2_toppic_prsm.csv
- A csv file containing identified proteoforms and their best PrSMs with a 1% proteoform level FDR: C:\toppic_tutorial\tutorial_1\st_1_ms2_toppic_proteoform.csv
- An xml file containing identified proteoforms and their best PrSMs with a 1% proteoform level FDR: C:\toppic_tutorial\tutorial_1\st_1_ms2_toppic_proteoform.xml
- A folder containing java script files of identified PrSMs with a 1% spectrum level FDR: C:\toppic_tutorial\tutorial_1\st_1_html\toppic_prsm_cutoff
- A folder containing java script files of identified PrSMs with a 1% proteoform level FDR: C:\toppic_tutorial\tutorial_1\st_1_html\toppic_proteoform_cutoff
- A folder containing html files for the visualization of identified PrSMs: C:\toppic_tutorial\tutorial_1\st_1_html\topview
In addition, the identifications reported for st_1_ms2.msalign and st_2_ms2.msalign are combined, and filtered by a 1% spectrum-level FDR and a 1% proteoform level FDR. The combined results are reported in the following files.
- A csv file containing combined PrSM identifications with a 1% spectrum level FDR: C:\toppic_tutorial\tutorial_1\combined_ms2_toppic_prsm.csv
- A csv file containing combined proteoform identifications and their best PrSMs with a 1% proteoform level FDR: C:\toppic_tutorial\tutorial_1\combined_ms2_toppic_proteoform.csv
- An xml file containing combined proteoform identifications and their best PrSMs with a 1% proteoform level FDR: C:\toppic_tutorial\tutorial_1\combined_ms2_toppic_proteoform.xml
- A folder containing java script files of combined PrSM identifications with a 1% spectrum level FDR: C:\toppic_tutorial\tutorial_1\combined_html\toppic_prsm_cutoff
- A folder containing java script files of combined PrSM identifications with a 1% proteoform level FDR: C:\toppic_tutorial\tutorial_1\combined_html\toppic_proteoform_cutoff
- A folder containing html files for the visualization of identified PrSMs: C:\toppic_tutorial\tutorial_1\combined_html\topview
In the analysis, C57 is selected as the fixed modification because proteins were reduced with dithiothreitol and alkylated with iodoacetamide before the MS experiment. When proteins are not reduced, C0 should be selected.
A shuffled decoy database is concatenated to the target database to estimate spectrum level and proteoform level FDRs. All identified PrSMs are first filtered by a 1% spectrum level FDR and the resulting PrSMs are reported in the file combined_ms2_toppic_prsm.csv. The proteoforms corresponding to the PrSMs are further filtered using a 1% proteoform level FDR and the resulting proteoforms and their corresponding best PrSMs are reported in the file combined_ms2_toppic_proteoform.csv. Microsoft Excel can be used to open these two files. To browse the PrSM identifications, go to the folder combined_html\topview and use Google Chrome (Windows IE and Firefox are not recommended) to open the file index.html.
The output files can be downloaded here.
4.6 Data analysis using the command line interface
4.6.1 Mass spectral deconvolution
We use topfd for top-down mass spectral deconvolution.
File location:
- Executable file: C:\toppic_tutorial\toppic\topfd.exe
- Input mzML file: C:\toppic_tutorial\tutorial_1\st_1.mzML
C:\toppic_tutorial\tutorial_1\st_2.mzML
Commands
cd c:\toppic_tutorial\tutorial_1
..\toppic\topfd st_*.mzML
4.6.2 Mass spectral identification using TopPIC
We use toppic to search the MS/MS spectra in st_1_ms2.msalign and st_2_ms2.msalign against the protein database uniprot-st.fasta to identify PrSMs.
File locations
- Executable file: C:\toppic_tutorial\toppic\toppic.exe
- Input protein database file: C:\toppic_tutorial\tutorial_1\unprot-st.fasta
- Input MS/MS msalign files: C:\toppic_tutorial\tutorial_1\st_1_ms2.msalign
C:\toppic_tutorial\tutorial_1\st_2_ms2.msalign - Input feature files: C:\toppic_tutorial\tutorial_1\st_1_ms1.feature
C:\toppic_tutorial\tutorial_1\st_1_ms2.feature
C:\toppic_tutorial\tutorial_1\st_2_ms1.feature
C:\toppic_tutorial\tutorial_1\st_2_ms2.feature
Commands
cd c:\toppic_tutorial\tutorial_1
..\toppic\toppic -f C57 -d -t FDR -T FDR -c combined uniprot-st.fasta st_*_ms2.msalign
5. Tutorial 2
We will use TopMG to analyze the data set st_1.raw described in Tutorial 1. TopMG is still in the development stage. Please let us know if you find any bugs in it..
5.1 Data set and preprocessing
The description of the data file and its preprocessing steps can be found in Sections 4.1 - 4.4. Click here to download the data files used in the analysis, save it in the folder C:\toppic_tutorial\tutorial_2\, and unzip it. It includes the following files.
- A S. typhrimurium protein database file: C:\toppic_tutorial\tutorial_2\uniprot-st.fasta
- A deconvoluted MS/MS data file: C:\toppic_tutorial\tutorial_2\st_1_ms2.msalign
- Two MS feature file: C:\toppic_tutorial\tutorial_2\st_1_ms1.feature
C:\toppic_tutorial\tutorial_2\st_1_ms2.feature - A text file containing a variable PTM: oxidation. C:\toppic_tutorial\tutorial_2\variable_ptms.txt
- A folder containing deconvoluted MS/MS spectra in the java script format. C:\toppic_tutorial\tutorial_2\st_1_html
- A folder containing a msalign file for MS1 spectra and a csv file for its mzRT features C:\toppic_tutorial\tutorial_2\st_1_file
5.2 Proteoform identification by TopMG
- Double click the executable file topmg_gui.exe in the folder C:\toppic_tutorial\toppic.
- Select C:\toppic_tutorial\tutorial_2\uniprot-st.fasta as the protein database file.
- Add C:\toppic_tutorial\tutorial_2\st_1_ms2.msalign as a mass spectrum data file.
- Select C:\toppic_tutorial\tutorial_2\variable_mods.txt as the file of variable PTMs.
- Select C57 as the fixed modification.
- Check the checkbox Decoy database.
- Select FDR as the spectrum level cutoff type.
- Set the spectrum level FDR cutoff to 0.05.
- Select FDR as the proteoform level cutoff type.
- Set the proteoform level FDR cutoff to 0.05.
- Click to the button "Start"
The screenshots of topmg_gui are shown below.
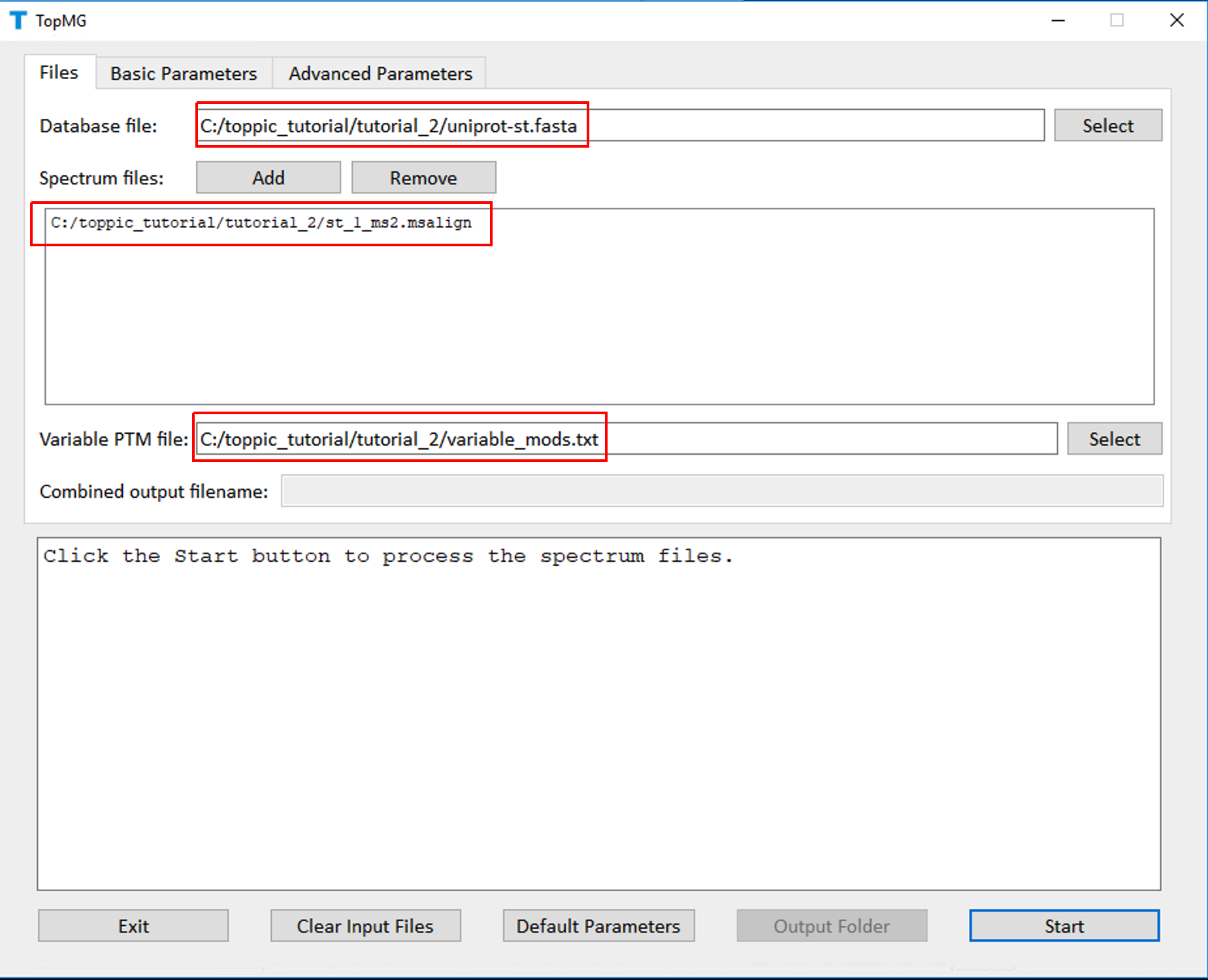
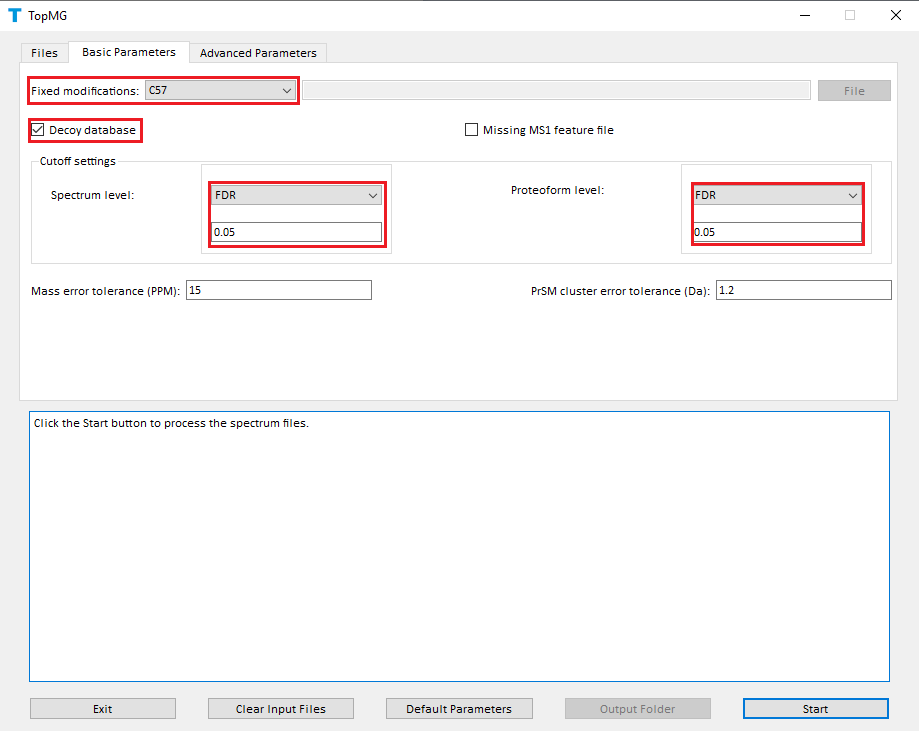
TopMG reports two cvs files, an xml file, and collections of html files for identified proteoforms.
- A csv file containing identified PrSMs with a 5% spectrum level FDR: C:\toppic_tutorial\tutorial_2\st_1_ms2_topmg_prsm.csv
- A csv file containing identified proteoforms and their best PrSMs with a 5% proteoform level FDR: C:\toppic_tutorial\tutorial_2\st_1_ms2_topmg_proteoform.csv
- An xml file containing identified proteoforms and their best PrSMs with a 5% proteoform level FDR: C:\toppic_tutorial\tutorial_2\st_1_ms2_topmg_proteoform.xml
- A folder containing java script files of identified PrSMs with a 5% spectrum level FDR: C:\toppic_tutorial\tutorial_2\st_1_html\topmg_prsm_cutoff
- A folder containing java script files of identified PrSMs with a 5% proteoform level FDR: C:\toppic_tutorial\tutorial_2\st_1_html\topmg_proteoform_cutoff
- A folder containing html files for the visualization of identified PrSMs: C:\toppic_tutorial\tutorial_1\st_1_html\topview
The output files can be downloaded here.
To browse the PrSM identifications, go to the folder st_1_html\topview and use Google Chrome (Windows IE and Firefox are not recommended) to open the file index.html.
5.3 Data analysis using the command line interface
File locations
- Executable file: C:\toppic_tutorial\toppic\topmg.exe
- Input protein database file: C:\toppic_tutorial\tutorial_2\unprot-st.fasta
- Input MS/MS msalign file: C:\toppic_tutorial\tutorial_2\st_1_ms2.msalign
- MS feature files: C:\toppic_tutorial\tutorial_2\st_1_ms1.feature
C:\toppic_tutorial\tutorial_2\st_1_ms2.feature - Variable PTM list: C:\toppic_tutorial\tutorial_2\variable_mods.txt
Commands
cd c:\toppic_tutorial\tutorial_2
..\toppic\topmg -f C57 -d -t FDR -v 0.05 -T FDR -V 0.05 -i variable_mods.txt uniprot-st.fasta st_1_ms2.msalign
6. Tutorial 3
We will use TopPIC and TopDiff to compare the abundance of proteoforms and find differentially expressed proteoforms using two MS data files of Escherichia coli cells (ecoli_1.raw and ecoli_2.raw).
In the MS experiment, the protein extract of E. coli was reduced with dithiothreitol and alkylated with iodoacetamide. The protein mixture was separated by capillary zone electrophoresis and analyzed by an LTQ-Orbitrap mass spectrometer (Thermo Fisher Scientific). Technical duplicates were generated for testing proteoform quantification in two runs of the same sample.
6.1 Data set and preprocessing
The raw data files were processed following the steps found in Sections 4.1 - 4.4. Click here to download the data files used in the analysis, save it in the folder C:\toppic_tutorial\tutorial_3\, and unzip it. It includes the following files.
- E.coli protein database file: C:\toppic_tutorial\tutorial_3\uniprot-ecoli.fasta
- Two deconvoluted MS/MS data files: C:\toppic_tutorial\tutorial_3\ecoli_1_ms2.msalign
C:\toppic_tutorial\tutorial_3\ecoli_2_ms2.msalign - Four deconvoluted MS/MS feature files: C:\toppic_tutorial\tutorial_3\ecoli_1_ms1.feature
C:\toppic_tutorial\tutorial_3\ecoli_2_ms1.feature
C:\toppic_tutorial\tutorial_3\ecoli_1_ms2.feature
C:\toppic_tutorial\tutorial_3\ecoli_2_ms2.feature - Two folders containing deconvoluted MS/MS spectra in the java script format. C:\toppic_tutorial\tutorial_3\ecoli_1_html
C:\toppic_tutorial\tutorial_3\ecoli_2_html - Two folders containing a msalign file for MS1 spectra and a csv file for its mzRT features C:\toppic_tutorial\tutorial_3\ecoli_1_file
C:\toppic_tutorial\tutorial_3\ecoli_2_file
6.2 Mass spectral identification using TopPIC
We use toppic_gui to search the MS/MS spectra in ecoli_1_ms2.msalign and ecoli_2_ms2.msalign against the protein database uniprot-ecoli.fasta to identify PrSMs.
- Double click the executable file toppic_gui.exe in the folder C:\toppic_tutorial\toppic.
- Select C:\toppic_tutorial\tutorial_3\uniprot-ecoli.fasta as the protein database file.
- Add C:\toppic_tutorial\tutorial_3\ecoli_1_ms2.msalign and C:\toppic_tutorial\tutorial_3\ecoli_2_ms2.msalign as mass spectrum data files.
- Select C57 as the fixed modification.
- Check the checkbox Decoy database.
- Select FDR as the spectrum level cutoff type.
- Select FDR as the proteoform level cutoff type.
- Click to the button "Start"
The screenshots of toppic_gui are shown below.
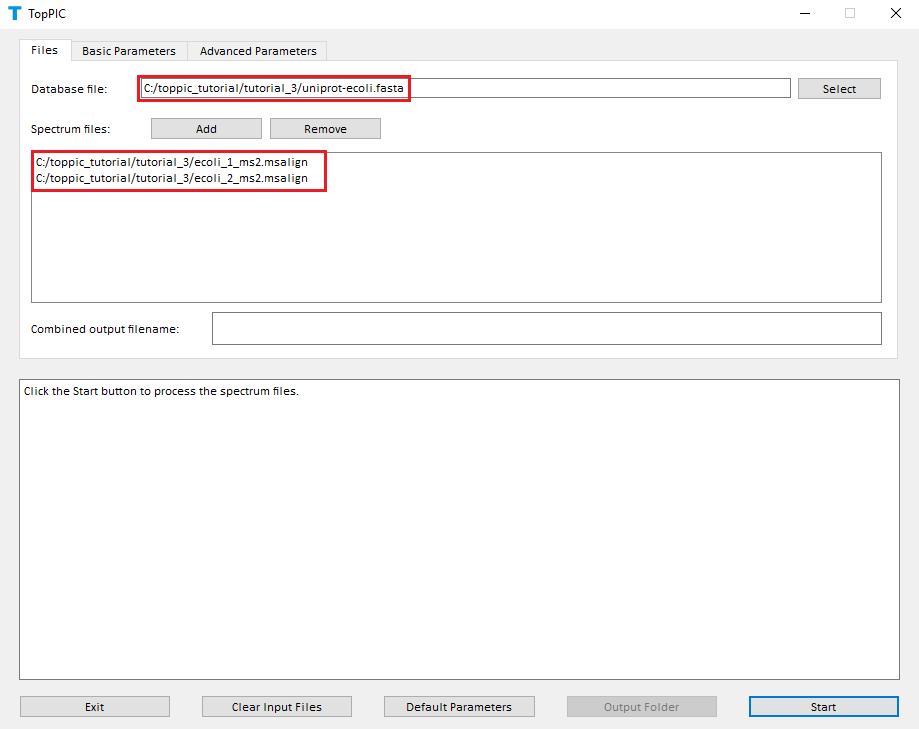
For each input msalign file, TopPIC reports two csv files, an xml file, and collections of html files for identified proteoforms. As a result, the output files for ecoli_1_ms2.msalign, ecoli_2_ms2.msalign are
- Two csv files containing identified PrSMs with a 1% spectrum level FDR: C:\toppic_tutorial\tutorial_3\ecoli_1_ms2_toppic_prsm.csv
C:\toppic_tutorial\tutorial_3\ecoli_2_ms2_toppic_prsm.csv - Two csv files containing identified proteoforms and their best PrSMs with a 1% proteoform level FDR: C:\toppic_tutorial\tutorial_3\ecoli_1_ms2_toppic_proteoform.csv
C:\toppic_tutorial\tutorial_3\ecoli_2_ms2_toppic_proteoform.csv - Two xml files containing identified proteoforms and their best PrSMs with a 1% proteoform level FDR: C:\toppic_tutorial\tutorial_3\ecoli_1_ms2_toppic_proteoform.xml
C:\toppic_tutorial\tutorial_3\ecoli_2_ms2_toppic_proteoform.xml - Two folders containing java script files of identified PrSMs with a 1% spectrum level FDR: C:\toppic_tutorial\tutorial_3\ecoli_1_html\toppic_prsm_cutoff
C:\toppic_tutorial\tutorial_3\ecoli_2_html\toppic_prsm_cutoff - Two folders containing java script files of identified PrSMs with a 1% proteoform level FDR: C:\toppic_tutorial\tutorial_3\ecoli_1_html\toppic_proteoform_cutoff
C:\toppic_tutorial\tutorial_3\ecoli_2_html\toppic_proteoform_cutoff - Two folders containing html files for the visualization of identified PrSMs: C:\toppic_tutorial\tutorial_3\ecoli_1_html\topview
C:\toppic_tutorial\tutorial_3\ecoli_2_html\topview
The output files can be downloaded here.
6.3 Proteoform abundance comparison by TopDiff
- Double click the executable file topdiff_gui.exe in the folder C:\toppic_tutorial\toppic.
- Select C:\toppic_tutorial\tutorial_3\uniprot-ecoli.fasta as the protein database file.
- Add C:\toppic_tutorial\tutorial_3\ecoli_1_ms2.msalign and C:\toppic_tutorial\tutorial_3\ecoli_2_ms2.msalign as a mass spectrum data file.
- Select C57 as the fixed modification.
- Click to the button "Start"
The screenshots of topdiff_gui are shown below.

TopDiff reports one csv file for identified proteoforms with their abundances in the input mass spectrum data
C:\toppic_tutorial\tutorial_3\sample_diff.csv
The output file can be downloaded here.
6.4 Data analysis using the command line interface
6.4.1 Mass spectral identification by TopPIC
File locations
- Executable file: C:\toppic_tutorial\toppic\toppic.exe
- Input protein database file: C:\toppic_tutorial\tutorial_3\unprot-ecoli.fasta
- Input MS/MS msalign files: C:\toppic_tutorial\tutorial_3\ecoli_1_ms2.msalign
C:\toppic_tutorial\tutorial_3\ecoli_2_ms2.msalign - MS feature files: C:\toppic_tutorial\tutorial_3\ecoli_1_ms1.feature
C:\toppic_tutorial\tutorial_3\ecoli_2_ms1.feature
C:\toppic_tutorial\tutorial_3\ecoli_1_ms2.feature
C:\toppic_tutorial\tutorial_3\ecoli_2_ms2.feature
Commands
cd c:\toppic_tutorial\tutorial_3
..\toppic\toppic -f C57 -d -t FDR -T FDR uniprot-ecoli.fasta ecoli_*_ms2.msalign
6.4.2 Comparing proteoform abundances using TopDiff
File locations
- Executable file: C:\toppic_tutorial\toppic\topdiff.exe
- Input protein database file: C:\toppic_tutorial\tutorial_3\unprot-ecoli.fasta
- Input MS/MS msalign files: C:\toppic_tutorial\tutorial_3\ecoli_1_ms2.msalign
C:\toppic_tutorial\tutorial_3\ecoli_2_ms2.msalign - Input proteoform identification files: C:\toppic_tutorial\tutorial_3\ecoli_1_ms2_toppic_proteoform.xml
C:\toppic_tutorial\tutorial_3\ecoli_2_ms2_toppic_proteoform.xml
Commands
cd c:\toppic_tutorial\tutorial_3
..\toppic\topdiff -f C57 unprot-ecoli.fasta ecoli_1_ms2.msalign ecoli_2_ms2.msalign